Actualité
13
mai 2026
Découvrez les projets qui ont besoin de vous
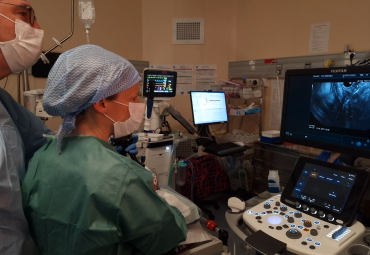
Le catalogue de projets du Fonds de dotation du CHU Grenoble Alpes est disponible dès maintenant. Consultez le à tout moment pour découvrir rapidement...
Lire la suite